Deficiencia de AADC
La deficiencia de L-aminoácido aromático descarboxilasa (AADC)
es un trastorno autosómico, recesivo, poco frecuente que afecta a la síntesis de neurotransmisores. Cursa con una amplia variedad de síntomas, que aparecen en los primeros años vida como disfunción motora y disautonomía que afectan a las actividades cotidianas y retraso del desarrollo.
Actualmente se conocen unas 100 variantes patogénicas en homocigosis o heterocigosis compuesta en pacientes con deficiencia de AADC.3,4,7,8
La variante más frecuente es la IVS6+4A>T; sin embargo, esta mutación está presente en menos de la mitad de los casos confirmados de deficiencia de AADC.2,3
No existe una relación clara entre la gravedad de la deficiencia de AADC y las variantes específicas conocidas; la mayoría de las variantes dan lugar a un cuadro clínico inicial variable.

La ausencia de actividad de la enzima AADC inhibe la síntesis de los neurotransmisores monoamina y causa los principales signos y síntomas de la deficiencia de AADC3
La ausencia de actividad de la enzima AADC da lugar a la acumulación de precursores de la serotonina y la dopamina (5-HTP, L-DOPA, 3-OMD), así como a la disminución de los niveles de los metabolitos de la serotonina y las catecolaminas (5-HIAA, HVA y MHPG).2
Esto produce una grave deficiencia de dopamina, serotonina y otras catecolaminas como noradrenalina y adrenalina.2,3
La ausencia de estos neurotransmisores inhibe la señalización neuronal en el sistema nervioso central, que afecta al desarrollo motor, a la conducta y a la función autonómica.3
La disminución de los niveles de los neurotransmisores contribuye a la aparición de los síntomas que experimentan los pacientes con deficiencia de AADC.6
La deficiencia de AADC cursa con una amplia variedad de manifestaciones clínicas.3,6
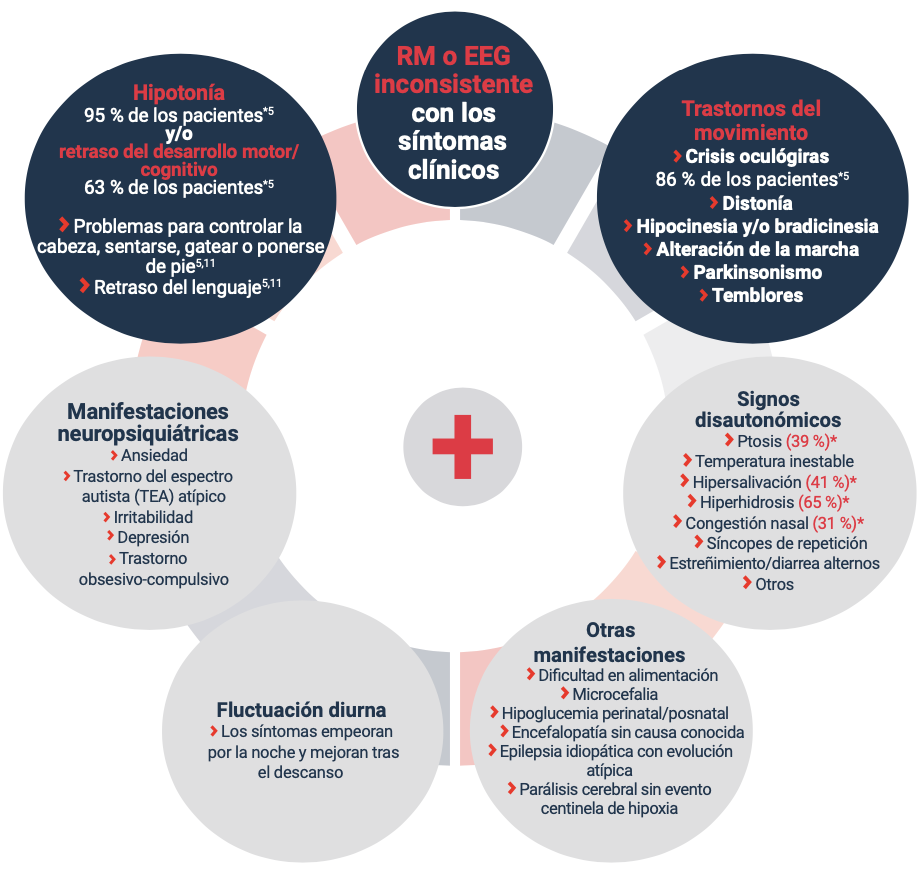
Los pacientes con deficiencia de AADC presentan un amplio espectro de fenotipos con disfunción motora, retraso del desarrollo y dificultad para alcanzar los principales hitos del desarrollo.2-9 Los pacientes con deficiencia de AADC también pueden presentar disautonomía.2,3
Algunos síntomas como hipotonía, crisis oculógiras, retraso del desarrollo motor y signos disautonómicos se asocian de manera sistemática a la deficiencia de AADC.3 No obstante, estos síntomas pueden ser de leves a graves, lo que explica la gran variabilidad de la presentación clínica.3 Los pacientes con las formas más graves de la deficiencia de AADC pueden no llegar a alcanzar nunca los hitos del desarrollo.9
Revisión bibliográfica de 117 pacientes con deficiencia de AADC en los que se describen las características de los pacientes en caso de estar disponibles2
Presentación clínica en pacientes con deficiencia de AADC
Los síntomas de la deficiencia de AADC suelen aparecer a una edad temprana; según las guías de consenso publicadas:2,3
- Los síntomas suelen aparecer el primer año de vida y la media de edad de los pacientes que muestran síntomas es 2,7 meses (n=68)
- A pesar de esta edad de aparición de los síntomas tan temprana, la media de edad en el diagnóstico es de 3,5 años *(n=87)
La deficiencia de AADC puede cursar con signos y síntomas similares a los de otras enfermedades neurológicas (ej. parálisis cerebral, epilepsia) y algunos trastornos de la conducta3
Los signos y síntomas de la deficiencia de AADC pueden coincidir con los de otras enfermedades.3

La deficiencia de AADC se ha documentado en todo el mundo1 y afecta a pacientes de diferentes orígenes como:2,3
- Asiáticos
- Caucásicos
- Árabes
- Iraníes
- Judíos
El término prevalencia en el nacimiento o prevalencia en recién nacidos se
prefiere al término
incidencia, que es el número de casos nuevos de la enfermedad
en un momento
determinado. La prevalencia es el número de personas vivas que padecen
una
enfermedad en un momento dado y es un término más preciso que se
utiliza para las
enfermedades genéticas en las que el genotipo está presente en el
nacimiento.17
Al igual que ocurre con otras muchas enfermedades raras que están
infradiagnosticadas, resulta
difícil calcular la prevalencia de la deficiencia de AADC. La estimación de la
prevalencia en el
nacimiento varía entre 1:32.000 y 1:182.000, dependiendo de la zona
geográfica.3,18,19

Se deben realizar pruebas a aquellos pacientes que presenten los principales signos y síntomas de la deficiencia de AADC para determinar la presencia de un trastorno de los neurotransmisores.
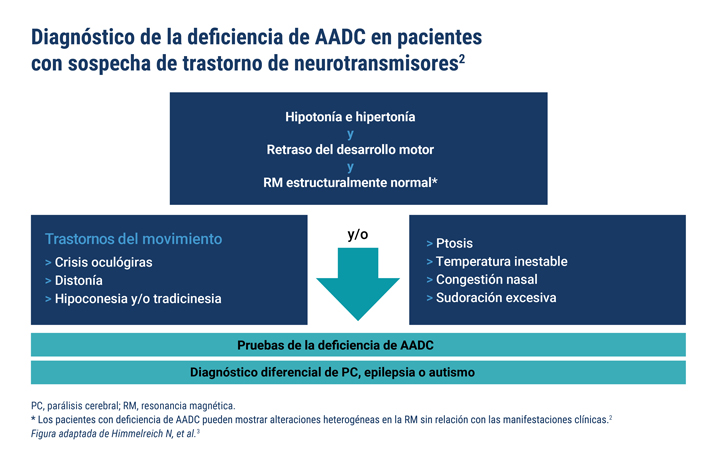
Las 3 pruebas diagnósticas básicas para confirmar la deficiencia de AADC son:2,3
-
Pruebas genéticas. Los pacientes con deficiencia de AADC tienen mutaciones en el gen DDC.
- Las guías de consenso actuales recomiendan realizar
una prueba genética para confirmar el
diagnóstico de deficiencia de AADC2.
- Las guías de consenso actuales recomiendan realizar
una prueba genética para confirmar el
diagnóstico de deficiencia de AADC2.
- Estudio de los metabolitos de los neurotransmisores en el líquido
cefalorraquídeo (LCR).
Resultados que cabe esperar en pacientes con deficiencia de AADC:
- Aumento de la concentración de L-DOPA, 3-OMD y 5-HTP
- Disminución de la concentración de HVA, 5-HIAA y MHPG
- Niveles normales de pterinas
- Estudio de la actividad enzimática en el plasma: en los pacientes con deficiencia de AADC se observa una actividad reducida de la enzima AADC en el plasma

De las 3 pruebas básicas, 2 tienen que ser positivas para confirmar el diagnóstico de la deficiencia de AADC2.
Existen otras pruebas para detectar la deficiencia de AADC, como el análisis de la concentración de 3-OMD en sangre o plasma y el análisis de ácidos orgánicos en orina2,19–23
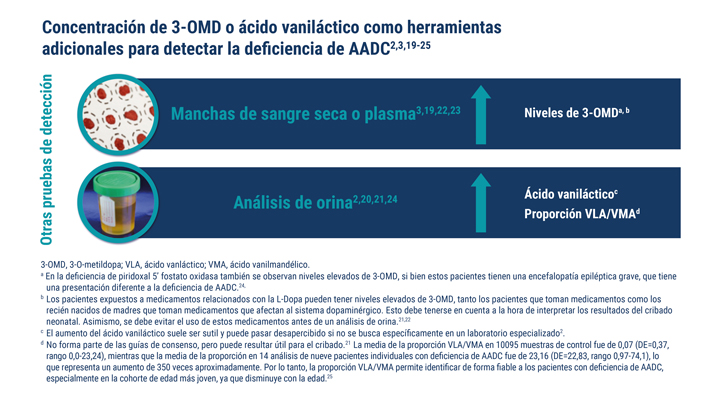
El análisis de muestras de sangre seca para determinar la concentración de 3-OMD es una prueba sencilla, rápida y mínimamente invasiva que puede utilizarse para confirmar la presencia de deficiencia de AADC.5,19,22
Podría incluirse en el cribado neonatal19,22 para favorecer el diagnóstico precoz de la deficiencia de AADC y reducir así el retraso en el diagnóstico de la enfermedad.2,19
Si la concentración de 3-OMD o de ácido vaniláctico es elevada, se deben realizar las pruebas básicas para confirmar el diagnóstico (estudio de la actividad en el plasma, análisis del LCR y pruebas genéticas).2
Los síntomas de la deficiencia de L-aminoácido aromático descarboxilasa (AADC) coinciden notablemente con los observados en otras enfermedades neurológicas, lo que puede llevar a un diagnóstico erróneo. En las siguientes infografías se muestran las manifestaciones clínicas de la deficiencia de AADC y de otras enfermedades solapantes más frecuentes como parálisis cerebral, epilepsia, trastornos neuromusculares y mitocondriopatías, así como los principales rasgos característicos que nos permitirán obtener un diagnóstico preciso y oportuno2,6,26-28.
Parálisis cerebral
La parálisis cerebral (PC) describe un grupo de trastornos permanentes del desarrollo neuromotor que causan limitaciones en la actividad. Se atribuyen a alteraciones no progresivas ocurridas en el desarrollo cerebral del feto o del lactante, acompañadas de retraso del desarrollo.29,30 Es la discapacidad física más frecuente en niños, con una prevalencia de entre 1,7 y 3,1 casos por cada 1.000 habitantes en los países desarrollados.31
Existen diferentes tipos de PC, que se clasifican según la zona del sistema nervioso central afectada y las manifestaciones clínicas:32,33
- La parálisis cerebral espástica es el tipo de parálisis cerebral más común y representa más del 70 % de los casos. Los síntomas son rigidez y espasticidad de los músculos, movimientos anormales, dificultades para controlar y coordinar los movimientos musculares y alteraciones de los movimientos de labios, lengua y paladar con disartria y disfagia. Por consiguiente, los pacientes no llegan a alcanzar hitos normales del desarrollo como sentarse, gatear y caminar.32,33
- La parálisis cerebral atetósica o discinética representa aproximadamente un 20 % de los casos y se caracteriza por movimientos lentos y ondulatorios (atetosis), movimientos repetitivos y de torsión (distonía) y movimientos irregulares e imprevisibles (corea), de lentos a rápidos y que pueden ir acompañados de dolor. Los movimientos tienden a aumentar con la tensión emocional y pueden desaparecer durante el sueño.32,33
- La parálisis cerebral atáxica se da en menos del 5 % de los casos. Los síntomas son debilidad, falta de coordinación, temblores y movimientos involuntarios, equilibrio inestable y dificultad para realizar movimientos rápidos o precisos.32,33
- La parálisis cerebral mixta es un trastorno frecuente en el que los pacientes presentan diferentes síntomas, si bien en general se caracteriza por la presencia de espasticidad y atetosis.32,33
Muchas de las manifestaciones de la PC durante la infancia son similares a las observadas en pacientes con trastornos de los neurotransmisores monoamina como la deficiencia de AADC, lo que a menudo puede llevar a un diagnóstico y tratamiento erróneo de PC.27 Por consiguiente, el término "enfermedad imitadora de la PC" se utiliza para describir una serie de trastornos neurogenéticos que se manifiestan de forma similar en la primera infancia.30
Los principales síntomas de la deficiencia de AADC que normalmente no se observan en la PC son las crisis oculógiras y la fluctuación diurna de los síntomas motores. En estos casos es necesario realizar las pruebas adecuadas, como un análisis de los neurotransmisores en el líquido cefalorraquídeo, para obtener un diagnóstico clínico preciso.30,34
Asimismo, los pacientes con enfermedades imitadoras de la PC muestran otros síntomas que difieren de la PC clásica y que pueden dar una pista a la hora de establecer el diagnóstico, como diferentes signos disautonómicos por ej. sudoración excesiva (diaforesis), temperatura inestable, secreción nasal y orofaríngea abundante, trastornos de los movimientos intestinales (dismotilidad gástrica) y trastornos del sueño.30
En pacientes pediátricos o adultos con un trastorno motor no diagnosticado compatible con PC, la prueba de primera línea consiste en una resonancia magnética cerebral. Los estudios de neuroimagen muestran claramente si la causa principal de los síntomas neurológicos es una malformación cerebral. Además de malformaciones cerebrales, estos estudios pueden mostrar resultados normales o lesiones específicas características de un trastorno genético o de un grupo de trastornos, que indiquen la necesidad de realizar más pruebas.30
Síntomas comunes y distintivos de la PC y la deficiencia de AADC*

*No se trata de una lista completa
Epilepsia
La epilepsia es un trastorno neurológico crónico que se caracteriza por crisis recurrentes e imprevisibles como consecuencia de una actividad neuronal anormal excesiva o síncrona en el cerebro.38,39
Es una de las enfermedades neurológicas más comunes en todo el mundo y afecta a unos 50 millones de personas de todas las edades.39
Las crisis epilépticas se dividen en tres categorías:40
- Las crisis generalizadas se inician en redes neuronales bilaterales en cualquier punto del cerebro y pueden ser de ausencia, tónico-clónicas generalizadas (TCG), mioclónicas y atónicas.
- Las crisis focales se originan en redes neuronales localizadas en una determinada región de un hemisferio cerebral y su manifestación clínica depende del área del córtex afectada.
- Los espasmos epilépticos se caracterizan por un movimiento de extensión o flexión súbita de las extremidades que se mantienen durante varios segundos y luego se repiten.
Los episodios paroxísticos que a menudo experimentan los pacientes con deficiencia de AADC, como las crisis oculógiras, postura tónica o distónica de las extremidades, mioclonía y corea, pueden confundirse con crisis epilépticas y, por consiguiente, diagnosticarse erróneamente como tales.2,6,41-43 Por esta razón, antes de confirmar el diagnóstico de deficiencia de AADC, los pacientes reciben múltiples tratamientos con fármacos antiepilépticos sin que se observe ninguna respuesta.43 En particular, los resultados del encefalograma (EEG) de los pacientes con deficiencia de AADC y los pacientes con epilepsia suelen diferir, por lo que es fundamental realizar un análisis del EEG ictal para distinguir entre los pacientes que experimentan episodios paroxísticos y los pacientes con epilepsia.41
Las crisis epilépticas son poco frecuentes en pacientes con deficiencia de AADC,5 si bien se han descrito algunos casos.10,41,44-46 Sobre la base de un número limitado de casos clínicos publicados, las crisis epilépticas tienden a producirse en los primeros años de vida, no son intensas, suelen ser TCG o crisis focales complejas y, una vez documentadas, se controlan bien con la administración de fármacos antiepilépticos convencionales.10,41,44-4641
Diferenciar las crisis epilépticas de los episodios paroxísticos no epilépticos mediante un análisis de EEG es fundamental para no diagnosticar erróneamente la deficiencia de AADC como epilepsia y garantizar que los pacientes reciban el tratamiento adecuado.41
Signos diferenciadores de las crisis epilépticas y la deficiencia de AADC

Trastornos Neuromusculares
Los trastornos neuromusculares son un amplio conjunto de enfermedades genéticas heterogéneas que afectan a los músculos o al sistema nervioso periférico.49-51 A continuación se enumeran algunos tipos de trastornos neuromusculares:49,52
- Esclerosis lateral amiotrófica (ELA)
- Enfermedad de Charcot-Marie-Tooth
- Esclerosis múltiple
- Distrofia muscular
- Miastenia grave
- Miopatía
- Miositis (polimiositis y dermatomiositis)
- Neuropatía periférica
- Atrofia muscular espinal
El problema puede tener su origen en los cuerpos celulares (ELA o en las ganglionopatías sensoriales), en los axones (neuropatías periféricas axonales o plexopatías braquiales), en las células de Schwann (polirradiculoneuropatía desmielinizante inflamatoria crónica), en las uniones neuromusculares (miastenia gravis o síndrome miasténico de Lambert-Eaton), en el tejido muscular (miopatía inflamatoria o distrofia muscular) o en una combinación de los anteriores.53
Los trastornos neuromusculares varían en cuanto a la evolución y la gravedad de la enfermedad54, en función de la región del cuerpo afectada, si bien los síntomas más frecuentes son debilidad muscular, alteración o deterioro de la marcha, contracturas articulares, deformidades óseas, alteración de la percepción sensorial (neuropatías) e insuficiencia respiratoria.49 Los pacientes con función muscular normal intermedia o alteraciones neuromusculares persistentes pueden experimentar mialgia, rabdomiólisis, debilidad y fatigabilidad.49
Teniendo en cuenta que el 50% de los pacientes con deficiencia de AADC presentan trastornos del movimiento (hipocinesia, corea, distonía, balismo, discinesia, temblor, mioclonía), la deficiencia de AADC puede confundirse con diferentes trastornos neuromusculares, como la miastenia gravis congénita, lo que puede ocasionar un diagnóstico erróneo.27,28
Síntomas comunes y distintivos de los trastornos neuromusculares y la deficiencia de AADC

Mitocondriopatías
Las mitocondriopatías son un grupo heterogéneo de enfermedades que se originan por la disfunción de la cadena respiratoria mitocondrial.56 Se calcula que la prevalencia de las mitocondriopatías es de al menos 20 de cada 100.000 habitantes.57
Las manifestaciones más frecuentes de las mitocondriopatías son ptosis, oftalmoplejía externa, miopatía proximal e intolerancia al ejercicio, cardiomiopatía, sordera neurosensorial, atrofia óptica, retinopatía pigmentaria y diabetes mellitus.56
Principales manifestaciones clínicas de las mitocondriopatías56

CPEO, oftalmoplejía externa progresiva crónica; LCR, líquido cefalorraquídeo; LHON, neuropatía óptica hereditaria de Leber; MELAS, encefalomiopatía mitocondrial con acidosis láctica y episodios similares a un accidente cerebrovascular; MEMSA, epilepsia mioclónica con ataxia sensorial; MERRF, epilepsia mioclónica con fibras rojas rasgadas; MIRAS, síndrome de ataxia mitocondrial recesiva; NARP, debilidad neurogénica con ataxia y retinitis pigmentaria; SANDO, neuropatía atáxica sensitiva-disartria-oftalmoplejía; SCAE, ataxia espinocerebelosa con epilepsia; KSS, síndrome de Kearns-Sayre; LIMM, miopatía mitocondrial letal del lactante; SNA, síndrome de neuropatía por ataxia.
*También denominada MIRAS y SCAE.
En lactantes, las manifestaciones clínicas como el síndrome rígido hipocinético y la distonía generalizada pueden confundirse con las observadas en la deficiencia de AADC y otros trastornos pediátricos de los neurotransmisores, lo que dificulta el diagnóstico basado únicamente en la presentación clínica.58 La presencia de crisis oculógiras puede ayudar a distinguir la deficiencia de AADC de una mitocondriopatía, sin embargo, no es suficiente para confirmar el diagnóstico ya que las crisis oculógiras también se observan en algunas mitocondriopatías.59 Por lo tanto, es necesario realizar pruebas genéticas moleculares para confirmar o descartar el diagnóstico de deficiencia de AADC.2
Síntomas comunes y distintivos de las mitocondriopatías y la deficiencia de AADC

*Las crisis convulsivas son poco frecuentes en la deficiencia de AADC, aunque se han observado algunos casos.
†Si bien se han observado casos de crisis oculógiras, estas no son típicas de las mitocondriopatías.
El proyecto RAINBOW es un programa de identificación y diagnóstico precoz de la deficiencia de AADC en España.
Existen 3 centros de referencia para el diagnóstico de esta patología en España, a los cuales se les puede solicitar el kit diagnóstico que enviarán de manera gratuita a todos aquellos médicos interesados con algún paciente con una clínica altamente sugestiva que lo soliciten.

Se recomienda realizar las pruebas diagnósticas para la deficiencia de AADC si el paciente cumple con las siguientes características:
En los pacientes con trastornos de los neurotransmisores, como la deficiencia de AADC, los síntomas pueden ser inespecíficos por lo que, a menudo, el diagnóstico se retrasa.61 Como consecuencia, los pacientes y los cuidadores recorren un largo camino hasta el diagnóstico y, a menudo, reciben diagnósticos erróneos, ej. parálisis cerebral, miastenia gravis o trastornos convulsivos, antes de dar con el diagnóstico correcto.61 Estos errores o retrasos en el diagnóstico pueden hacer que el estado del paciente empeore.61
Los pacientes pueden experimentar otras complicaciones como problemas cardíacos y ortopédicos.2 Además del retraso del crecimiento, estos pacientes son más propensos a sufrir infecciones, normalmente como consecuencia de complicaciones derivadas de los problemas de alimentación y deglución, la movilidad reducida y los ingresos recurrentes.2
La continua necesidad de asistencia física y los problemas de conducta de los pacientes con deficiencia de AADC representan una importante carga para los cuidadores2,3
Si bien no se dispone de mucha información sobre la calidad de vida de los cuidadores de pacientes con deficiencia de AADC, podemos hacernos una idea a partir de las experiencias de los cuidadores de otras enfermedades neurológicas pediátricas con síntomas similares.2
- Los resultados de las encuestas realizadas a cuidadores de pacientes con parálisis cerebral muestran que estos tienden a tener mayores niveles de estrés y depresión y una calidad de vida inferior a la de los padres de niños sanos.62 Los factores mencionados con mayor frecuencia como causa de estos efectos son los problemas cognitivos y conductuales del niño, el bajo nivel de autoeficacia del cuidador y la falta de apoyo social.62
En el caso de las enfermedades de los neurotransmisores, como la deficiencia de AADC, los errores frecuentes y el retraso en el diagnóstico son una causa de estrés para los cuidadores y suponen una carga económica considerable para el sistema sanitario.61
En muchos pacientes con deficiencia de AADC, los retrasos del desarrollo, las deficiencias motoras profundas y otras comorbilidades psiquiátricas o del neurodesarrollo que requieren cuidados de por vida, como ocurre en otras enfermedades crónicas como la epilepsia, suponen una pesada carga para los cuidadores y un deterioro de la calidad de vida.2,63,64
*Estudio retrospectivo, descriptivo y unicéntrico de pacientes diagnosticados de deficiencia de AADC en el Hospital Universitario Nacional de Taiwán entre 2004 y 2016

Manejo de la deficiencia de AADC: el panorama de
la
investigación
El profesor Thomas Opladen proporciona
información sobre el panorama
de la investigación en la deficiencia de AADC.
Revise los estudios de casos clínicos en esta página, guiados por los principales expertos, para reforzar su comprensión de la deficiencia de L-aminoácido aromático descarboxilasa (AADC). Las preguntas interactivas pondrán a prueba sus conocimientos y lo ayudarán a realizar diagnósticos precisos y oportunos.






Ver todos los vídeos sobre AADC-d
Los congresos, conferencias y reuniones internacionales ofrecen la oportunidad de compartir datos e impulsar la discusión que puede avanzar en el manejo de afecciones como la deficiencia de L-aminoácido aromático descarboxilasa (AADC).
Acceda nuestro contenido con información de los últimos congresos y reuniones de las principales sociedades científicas en la siguiente página web:

2. Wassenberg T, Molero-Luis M, Jeltsch K, Hoffmann GF, Assmann B, Blau N, et al. Consensus guideline for the diagnosis and treatment of aromatic l-amino acid decarboxylase (AADC) deficiency. Orphanet J Rare Dis. 2017;12(1):12. doi: 10.1186/s13023-016-0522-z. https://pubmed.ncbi.nlm.nih.gov/28100251/
3. Himmelreich N, Montioli R, Bertoldi M, Carducci C, Leuzzi V, Gemperle C, et al. Aromatic amino acid decarboxylase deficiency: Molecular and metabolic basis and therapeutic outlook. Mol Genet Metab. 2019;127(1):12-22. doi: 10.1016/j.ymgme.2019.03.009. https://pubmed.ncbi.nlm.nih.gov/30952622/
4. Dai L, Ding C, Fang F. A novel DDC gene deletion mutation in two Chinese mainland siblings with aromatic l-amino acid decarboxylase deficiency. Brain Dev. 2019;41(2):205-9. doi: 10.1016/j.braindev.2018.08.003. https://pubmed.ncbi.nlm.nih.gov/30144970/
5. Chen PW, Lee NC, Chien YH, Wu JY, Wang PC, Hwu WL. Diagnosis of aromatic L-amino acid decarboxylase deficiency by measuring 3-O-methyldopa concentrations in dried blood spots. Clin Chim Acta. 2014;431:19-22. doi: 10.1016/j.cca.2014.01.034. https://pubmed.ncbi.nlm.nih.gov/24513538/
6. Brun L, Ngu LH, Keng WT, Ch'ng GS, Choy YS, Hwu WL, et al. Clinical and biochemical features of aromatic L-amino acid decarboxylase deficiency. Neurology. 2010;75(1):64-71. doi: 10.1212/WNL.0b013e3181e620ae. Erratum in: Neurology. 2010;75(6):576. Dosage error in article text. https://pubmed.ncbi.nlm.nih.gov/20505134/
7. Monteleone B, Hyland K. Case report: discovery of 2 gene variants for aromatic L-amino acid decarboxylase deficiency in 2 African American siblings. BMC Neurol. 2020;20(1):12. doi: 10.1186/s12883-019-1596-8. https://pubmed.ncbi.nlm.nih.gov/31918669/
8. Micallef J, Stockler-Ipsiroglu S, van Karnebeek CD, Salvarinova-Zivkovic R, Horvath G. Recurrent Dystonic Crisis and Rhabdomyolysis Treated with Dantrolene in Two Patients with Aromatic L-Amino Acid Decarboxylase Deficiency. Neuropediatrics. 2020;51(3):229-32. doi: 10.1055/s-0039-3402010. https://pubmed.ncbi.nlm.nih.gov/31935764/
9. Hwu WL, Chien YH, Lee NC, Li MH. Natural History of Aromatic L-Amino Acid Decarboxylase Deficiency in Taiwan. JIMD Rep. 2018;40:1-6. doi: 10.1007/8904_2017_54. https://pubmed.ncbi.nlm.nih.gov/28856607/
10. Manegold C, Hoffmann GF, Degen I, Ikonomidou H, Knust A, Laass MW, et al. Aromatic L-amino acid decarboxylase deficiency: clinical features, drug therapy and follow-up. J Inherit Metab Dis. 2009;32(3):371-80. doi: 10.1007/s10545-009-1076-1. https://pubmed.ncbi.nlm.nih.gov/19172410/
11. Blackburn JS, Mink JW, Augustine EF. Pediatric movement disorders: Five new things. Neurol Clin Pract. 2012;2(4):311-8. doi: 10.1212/CPJ.0b013e318278bf06. https://pubmed.ncbi.nlm.nih.gov/23634375/
12. Hallman-Cooper JL, Rocha Cabrero F. Cerebral Palsy. [Updated 2021 Apr 28]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan. www.ncbi.nlm.nih.gov/books/NBK538147/ https://www.ncbi.nlm.nih.gov/books/NBK538147/
13. Menghi V, Bisulli F, Tinuper P, Nobili L. Sleep-related hypermotor epilepsy: prevalence, impact and management strategies. Nat Sci Sleep. 2018;10:317-26. doi: 10.2147/NSS.S152624. https://pubmed.ncbi.nlm.nih.gov/30349413/
14. Shibata M, Kato T, Yoshida T, Saito K, Awaya T, Heike T. Paroxysmal gaze deviations as the sole manifestation of occipital lobe epilepsy. Seizure. 2013;22(10):913-5. doi: 10.1016/j.seizure.2013.06.012. https://pubmed.ncbi.nlm.nih.gov/23870820/
15. DeFilippis M, Wagner KD. Treatment of Autism Spectrum Disorder in Children and Adolescents. Psychopharmacol Bull. 2016;46(2):18-41. https://pubmed.ncbi.nlm.nih.gov/27738378/
16. Kim JS. Excessive crying: behavioral and emotional regulation disorder in infancy. Korean J Pediatr. 2011;54(6):229-33. doi: 10.3345/kjp.2011.54.6.229. https://pubmed.ncbi.nlm.nih.gov/21949516/
17. Verhaart IEC, Robertson A, Wilson IJ, Aartsma-Rus A, Cameron S, Jones CC, et al. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review. Orphanet J Rare Dis. 2017;12(1):124. doi: 10.1186/s13023-017-0671-8. https://pubmed.ncbi.nlm.nih.gov/28676062/
18. Hyland K, Reott M. Prevalence of Aromatic l-Amino Acid Decarboxylase Deficiency in At-Risk Populations. Pediatr Neurol. 2020;106:38-42. doi: 10.1016/j.pediatrneurol.2019.11.022. https://pubmed.ncbi.nlm.nih.gov/32111562/
19. Chien YH, Chen PW, Lee NC, Hsieh WS, Chiu PC, Hwu WL, et al. 3-O-methyldopa levels in newborns: Result of newborn screening for aromatic l-amino-acid decarboxylase deficiency. Mol Genet Metab. 2016;118(4):259-63. doi: 10.1016/j.ymgme.2016.05.011. https://pubmed.ncbi.nlm.nih.gov/27216367/
20. Hyland K, Clayton PT. Aromatic L-amino acid decarboxylase deficiency: diagnostic methodology. Clin Chem. 1992;38(12):2405-10. https://pubmed.ncbi.nlm.nih.gov/1281049/
21. Lee HC, Lai CK, Yau KC, Siu TS, Mak CM, Yuen YP, et al. Non-invasive urinary screening for aromatic L-amino acid decarboxylase deficiency in high-prevalence areas: a pilot study. Clin Chim Acta. 2012;413(1-2):126-30. doi: 10.1016/j.cca.2011.09.008. https://pubmed.ncbi.nlm.nih.gov/21963339/
22. Brennenstuhl H, Kohlmüller D, Gramer G, Garbade SF, Syrbe S, Feyh P, et al. High throughput newborn screening for aromatic ʟ-amino-acid decarboxylase deficiency by analysis of concentrations of 3-O-methyldopa from dried blood spots. J Inherit Metab Dis. 2020;43(3):602-10. doi: 10.1002/jimd.12208. https://pubmed.ncbi.nlm.nih.gov/31849064/
23. Atwal PS, Donti TR, Cardon AL, Bacino CA, Sun Q, Emrick L, et al. Aromatic L-amino acid decarboxylase deficiency diagnosed by clinical metabolomic profiling of plasma. Mol Genet Metab. 2015;115(2-3):91-4. doi: 10.1016/j.ymgme.2015.04.008. https://pubmed.ncbi.nlm.nih.gov/25956449/
24. Mills PB, Surtees RA, Champion MP, Beesley CE, Dalton N, Scambler PJ, et al. Neonatal epileptic encephalopathy caused by mutations in the PNPO gene encoding pyridox(am)ine 5'-phosphate oxidase. Hum Mol Genet. 2005;14(8):1077-86. doi: 10.1093/hmg/ddi120. https://pubmed.ncbi.nlm.nih.gov/15772097/
25. Brennenstuhl H, Garbade SF, Okun JG, Feyh P, Hoffmann GF, Langhans CD, et al. Semi-quantitative detection of a vanillactic acid/vanillylmandelic acid ratio in urine is a reliable diagnostic marker for aromatic L-amino acid decarboxylase deficiency. Mol Genet Metab. 2020;131(1-2):163-70. doi: 10.1016/j.ymgme.2020.07.001. https://pubmed.ncbi.nlm.nih.gov/32675002/
26. Krigger KW. Cerebral palsy: an overview.Am Fam Physician. 2006;73(1):91-100. https://pubmed.ncbi.nlm.nih.gov/16417071/
27. Ng J, Papandreou A, Heales SJ, Kurian MA. Monoamine neurotransmitter disorders — clinical advances and future Perspectives. Nat Rev Neurol. 2015;11(10):567-84. https://pubmed.ncbi.nlm.nih.gov/26392380/
28. Kurian MA, Dale RC. Movement disorders presenting in childhood. Continuum (Minneap Minn). 2016;22(4 Movement Disorders):1159-85.https://pubmed.ncbi.nlm.nih.gov/27495203/
29. Novak I, Morgan C, Adde L, Blackman J, Boyd RN, Brunstrom-Hernandez J, et al. Early, Accurate Diagnosis and Early Intervention in Cerebral Palsy: Advances in Diagnosis and Treatment. JAMA Pediatr. 2017;171(9):897-907. doi: 10.1001/jamapediatrics.2017.1689. Erratum in: JAMA Pediatr. 2017;171(9):919. https://pubmed.ncbi.nlm.nih.gov/28715518/
30. Pearson TS, Pons R, Ghaoui R, Sue CM. Genetic mimics of cerebral palsy. Mov Disord. 2019;34(5):625-36. doi: 10.1002/mds.27655. https://pubmed.ncbi.nlm.nih.gov/30913345/
31. Monbaliu E, Himmelmann K, Lin JP, Ortibus E, Bonouvrié L, Feys H, et al. Clinical presentation and management of dyskinetic cerebral palsy. Lancet Neurol. 2017;16(9):741-9. doi: 10.1016/S1474-4422(17)30252-1. https://pubmed.ncbi.nlm.nih.gov/28816119/
32. Cerebral Palsy Guidance. Cerebral Palsy Symptoms. Available from: https://www.cerebralpalsyguidance.com/cerebral-palsy/symptoms/.
33. Cerebral Palsy (CP) Syndromes. (n.d.). Merck Manuals Professional Edition. Available from: https://www.merckmanuals.com/professional/pediatrics/neurologic-disorders-in-children/cerebral-palsy-cp-syndromes.
34. Kurian MA, Gissen P, Smith M, Heales S Jr, Clayton PT. The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol. 2011;10(8):721-33. doi: 10.1016/S1474-4422(11)70141-7. https://pubmed.ncbi.nlm.nih.gov/21777827/
35. Zouvelou V, Yubero D, Apostolakopoulou L, Kokkinou E, Bilanakis M, Dalivigka Z, et al. The genetic etiology in cerebral palsy mimics: The results from a Greek tertiary care center. Eur J Paediatr Neurol. 2019;23(3):427-37. doi: 10.1016/j.ejpn.2019.02.001. https://pubmed.ncbi.nlm.nih.gov/30799092/
36. Gururaj AK, Sztriha L, Bener A, Dawodu A, Eapen V. Epilepsy in children with cerebral palsy. Seizure. 2003 Mar;12(2):110-4. doi: 10.1016/s1059131102002558. https://pubmed.ncbi.nlm.nih.gov/12566235/
37. Singhi P, Jagirdar S, Khandelwal N, Malhi P. Epilepsy in children with cerebral palsy. J Child Neurol. 2003;18(3):174-9. DOI: 10.1177/08830738030180030601 https://pubmed.ncbi.nlm.nih.gov/12731642/
38. Fisher RS, Cross JH, French JA, Higurashi N, Hirsch E, Jansen FE, et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017 Apr;58(4):522-530. doi: 10.1111/epi.13670. https://pubmed.ncbi.nlm.nih.gov/28276060/
39. World Health Organization. Epilepsy: a public health imperative. 2019. Available from: https://www.who.int/mental_health/neurology/epilepsy/report_2019/en/..
40. Stafstrom CE, Carmant L. Seizures and epilepsy: an overview for neuroscientists. Cold Spring Harb Perspect Med. 2015;5(6):a022426. doi: 10.1101/cshperspect.a022426. https://pubmed.ncbi.nlm.nih.gov/26033084/
41. Ito S, Nakayama T, Ide S, Ito Y, Oguni H, Goto Y, et al. Aromatic L-amino acid decarboxylase deficiency associated with epilepsy mimicking non-epileptic involuntary movements. Dev Med Child Neurol. 2008;50(11):876-8. doi: 10.1111/j.1469-8749.2008.03094.x. https://pubmed.ncbi.nlm.nih.gov/18754761/
42. Pons R, Ford B, Chiriboga CA, Clayton PT, Hinton V, Hyland K, et al. Aromatic L-amino acid decarboxylase deficiency: clinical features, treatment, and prognosis. Neurology. 2004;62(7):1058-65. doi: 10.1212/wnl.62.7.1058. https://pubmed.ncbi.nlm.nih.gov/15079002/
43. Lee W-T. Disorders of monoamine metabolism: inherited disorders frequently misdiagnosed as epilepsy. Epilepsy Seizure. 2010;3(1):147-53. doi: 10.3805/eands.3.147. https://pubmed.ncbi.nlm.nih.gov/21803516/
44. Swoboda KJ, Saul JP, McKenna CE, Speller NB, Hyland K. Aromatic L-amino acid decarboxylase deficiency: overview of clinical features and outcomes. Ann Neurol. 2003;54 Suppl 6:S49-55. doi: 10.1002/ana.10631. https://pubmed.ncbi.nlm.nih.gov/12891654/
45. Hsieh HJ, Lin SH, Liu HM. Visualisation of impaired dopamine biosynthesis in a case of aromatic L-amino acid decarboxylase deficiency by co-registered 18F-FDOPA PET and magnetic resonance imaging. Eur J Nucl Med Mol Imaging. 2005;32(4):517. doi: 10.1007/s00259-004-1618-6. https://pubmed.ncbi.nlm.nih.gov/15821966/
46. Anselm IA, Darras BT. Catecholamine toxicity in aromatic L-amino acid decarboxylase deficiency. Pediatr Neurol. 2006;35(2):142-4. doi: 10.1016/j.pediatrneurol.2006.01.008. https://pubmed.ncbi.nlm.nih.gov/16876014/
47. Pearson TS, Gilbert L, Opladen T, García-Cazorla A, Mastrangelo M, Leuzzi V, et al. AADC deficiency from infancy to adulthood: Symptoms and developmental outcome in an international cohort of 63 patients. J Inherit Metab Dis. 2020;43(5):1121-30. doi: 10.1002/jimd.12247. https://pubmed.ncbi.nlm.nih.gov/32369189/
48. Orphanet. Progressive myoclonic epilepsy with dystonia. Available from: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Expert=352596&lng=EN.
49. Dowling JJ, D Gonorazky H, Cohn RD, Campbell C. Treating pediatric neuromuscular disorders: The future is now. Am J Med Genet A. 2018;176(4):804-41. doi: 10.1002/ajmg.a.38418. https://pubmed.ncbi.nlm.nih.gov/28889642/
50. van Putten M, Hmeljak J, Aartsma-Rus A, Dowling JJ. Moving neuromuscular disorders research forward: from novel models to clinical studies. Dis Model Mech. 2020;13(2):dmm044370. doi: 10.1242/dmm.044370. https://pubmed.ncbi.nlm.nih.gov/32224497/
51. Mary P, Servais L, Vialle R. Neuromuscular diseases: Diagnosis and management. Orthop Traumatol Surg Res. 2018;104(1S):S89-S95. doi: 10.1016/j.otsr.2017.04.019. https://pubmed.ncbi.nlm.nih.gov/29196274/
52. Deenen JC, Horlings CG, Verschuuren JJ, Verbeek AL, van Engelen BG. The Epidemiology of Neuromuscular Disorders: A Comprehensive Overview of the Literature. J Neuromuscul Dis. 2015;2(1):73-85. https://pubmed.ncbi.nlm.nih.gov/28198707/
53. Morrison BM. Neuromuscular Diseases. Semin Neurol. 2016;36(5):409-18. doi: 10.1055/s-0036-1586263. https://pubmed.ncbi.nlm.nih.gov/27704495/
54. Sáez A, Acha B, Montero-Sánchez A, Rivas E, Escudero LM, Serrano C. Neuromuscular disease classification system. J Biomed Opt. 2013;18(6):066017. doi: 10.1117/1.JBO.18.6.066017. https://pubmed.ncbi.nlm.nih.gov/23804164/
55. Kaler J, Hussain A, Patel S, Majhi S. Neuromuscular Junction Disorders and Floppy Infant Syndrome: A Comprehensive Review. Cureus. 2020;12(2):e6922. doi: 10.7759/cureus.6922. https://pubmed.ncbi.nlm.nih.gov/32071826/
56. Adam MP, Ardinger HH, Pagon RA, et al (Editors). GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020.https://pubmed.ncbi.nlm.nih.gov/20301295/
57. Schaefer AM, Taylor RW, Turnbull DM, Chinnery PF. The epidemiology of mitochondrial disorders - past, present and future. Biochim Biophys Acta. 2004;1659(2-3):115-20. doi: 10.1016/j.bbabio.2004.09.005. https://pubmed.ncbi.nlm.nih.gov/15576042/
58. Marecos C, Ng J, Kurian MA. What is new for monoamine neurotransmitter disorders? J Inherit Metab Dis. 2014;37(4):619-26. doi: 10.1007/s10545-014-9697-4. https://pubmed.ncbi.nlm.nih.gov/24696406/
59. García-Cazorla A, Duarte S, Serrano M, Nascimento A, Ormazabal A, Carrilho I, et al. Mitochondrial diseases mimicking neurotransmitter defects. Mitochondrion. 2008;8(3):273-8. doi: 10.1016/j.mito.2008.05.001. https://pubmed.ncbi.nlm.nih.gov/18558519/
60. Gropman AL. Neuroimaging in mitochondrial disorders. Neurotherapeutics. 2013;10(2):273-85. doi: 10.1007/s13311-012-0161-6. https://pubmed.ncbi.nlm.nih.gov/23208728/
61. Opladen T, Cortès-Saladelafont E, Mastrangelo M, Horvath G, Pons R, Lopez-Laso E, et al; International Working Group on Neurotransmitter related disorders (iNTD). The International Working Group on Neurotransmitter related Disorders (iNTD): A worldwide research project focused on primary and secondary neurotransmitter disorders. Mol Genet Metab Rep. 2016;9:61-6. doi: 10.1016/j.ymgmr.2016.09.006. https://pubmed.ncbi.nlm.nih.gov/27830117/
62. Pousada M, Guillamón N, Hernández-Encuentra E, Muñoz E, Redolar D, Boixadós M, et al. Impact of Caring for a Child with Cerebral Palsy on the Quality of Life of Parents: A Systematic Review of the Literature. J Dev Phys Disabil 2013;25:545-77. doi.org/10.1007/s10882-013-9332-6. https://link.springer.com/article/10.1007/s10882-013-9332-6
63. Lai ST, Tan WY, Wo MC, Lim KS, Ahmad SB, Tan CT. Burden in caregivers of adults with epilepsy in Asian families. Seizure. 2019;71:132-9. doi: 10.1016/j.seizure.2019.07.008. https://pubmed.ncbi.nlm.nih.gov/31325820/
64. Landfeldt E, Lindgren P, Bell CF, Guglieri M, Straub V, Lochmüller H, et al. Quantifying the burden of caregiving in Duchenne muscular dystrophy. J Neurol. 2016;263(5):906-15. doi: 10.1007/s00415-016-8080-9. https://pubmed.ncbi.nlm.nih.gov/26964543/

Eventos virtuales sobre AADC-d



ABORDAJE DIAGNÓSTICO EN LAS ENFERMEDADES HEREDITARIAS DEL METABOLISMO DE LOS NEUROTRANSMISORES
-
Dr. Eduardo López Laso
Dr. Salvador Ibáñez Micó
Dra. Ángeles García-Cazorla
En el marco de la reunión SENEP-Live organizada por la Sociedad Española de Neurología Pediátrica (SENEP), el pasado 21 de septiembre de 2020...


Recursos y vídeos sobre AADC-d







Este sitio web está dirigido exclusivamente al personal sanitario facultado para preescribir o dispensar medicamentos. Si usted no es un profesional sanitario, le invitamos a que visite nuestra web para el público
Este sitio web está organizado y financiado por PTC Therapeutics Spain, S.L. El contenido de este sitio web y la información provista en el mismo son propiedad de PTC Therapeutics Spain, S.L. El contenido de este sitio web no está destinado a ser distribuido, duplicado, copiado o compartido, ya sea parcialmente o en su totalidad sin el permiso previo por escrito de PTC Therapeutics Spain, S.L.
©2025 PTC Therapeutics Spain, S.L. Todos los derechos reservados
ES-PTC-2022-009| Última modificación: Junio 2025